tRNAscan-SE input consists of DNA or RNA sequences in FASTA format. tRNA predictions are output in tabular, ACeDB, or an extended format including tRNA secondary structure information. tRNAscan-SE does no tRNA detection itself, but instead negotiates the flow of information between three independent tRNA prediction programs, performs some post-processing, and outputs the results (Figure 2.1).
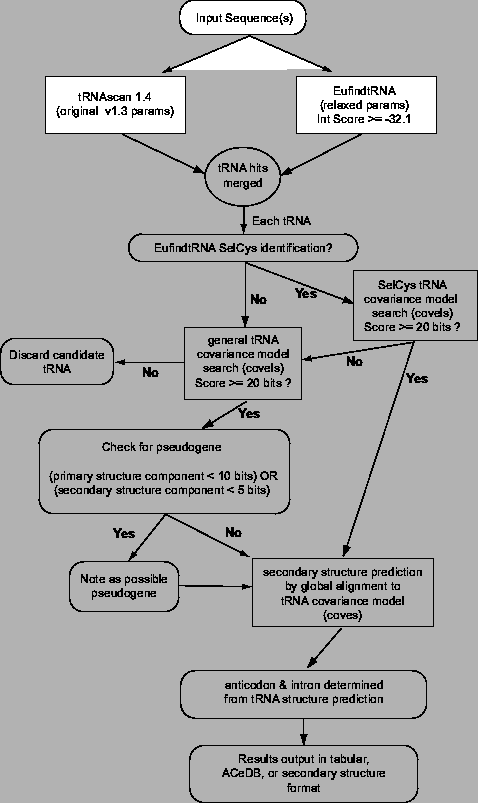 |
tRNAscan-SE works in three phases. In the first stage, it runs tRNAscan and the Pavesi algorithm on the input sequence. The first of these two programs is an optimized version of tRNAscan 1.3 [Fichant & Burks, 1991]. The other is an implementation of the Pavesi search algorithm [Pavesi et al., 1994] which we call EufindtRNA. Results from both programs are merged into one list of candidate tRNAs. Intron information from tRNAscan 1.3 is discarded because its intron predictions are typically unreliable. Analysis with the tRNA covariance model at a later stage (described below) allows non-ambiguous determination of intron boundaries.
In the second stage, tRNAscan-SE extracts the candidate subsequences and passes these segments to the covariance model search program covels [Eddy & Durbin, 1994]. Seven flanking nucleotides on both sides of the candidate tRNAs are included in the subsequence in case the tRNA was truncated by the initial prediction. The covels search program applies a tRNA covariance model (TRNA2.cm) that was made by structurally aligning 1415 tRNAs from the 1993 Sprinzl database [Steinberg et al., 1993]. 87 non-canonical ``group III'' sequences and 509 RNA sequences were removed from the complete 2011 sequence database as described in [Eddy & Durbin, 1994]. To improve intron prediction, intron sequences were manually inserted into the Sprinzl alignment for 38 intron-containing tRNAs of known genomic sequence.
Finally, tRNAscan-SE takes predicted tRNAs that have been confirmed with covels log odds scores of over 20.0 bits, trims the tRNA bounds to those predicted by covels, and runs the covariance model global structure alignment program coves [Eddy & Durbin, 1994] to get a secondary structure prediction. The tRNA isotype is predicted by identifying the anticodon within the coves secondary structure output. Introns are identified from this output as runs of five or more consecutive non-consensus nucleotides within the anticodon loop.
tRNAscan-SE uses heuristics to try to distinguish pseudogenes from true tRNAs, primarily on lack of tRNA-like secondary structure. A second tRNA covariance model (TRNA2ns.cm) was created from the same alignment, under the constraint that no secondary structure is conserved (this model is effectively just a sequence profile, or hidden Markov model). By subtracting a tRNA's similarity score to the primary structure-only model from that using the complete tRNA model, a secondary structure-only score is obtained. In Bayesian terms, this difference can be viewed as the evidence for the complete tRNA model, as opposed to a structure-less, sequence-only pseudogene model. We observed that tRNAs with low scores for either component of the total score were often pseudogenes. Thus, tRNAs are marked as likely pseudogenes if they have either a score of less than 10 bits for the primary sequence component of the total score, or a score of less than 5 bits for the secondary structure component of the total score. Selenocysteine tRNAs are not checked by these rules since they have atypical primary and secondary structure. Final tRNA predictions are then saved in tabular, ACeDB, or secondary structure output format.