


The traditional method used in computational biology to demonstrate the superiority of one technique to another is to compare them on a biologically interesting search or alignment problem. Many of the regularizers in Section 5 have been validated in this way [9,4,17]. This sort of anecdotal evidence is valuable for establishing the usefulness of techniques in real biological problems, but is very difficult to quantify.
In this paper, regularizers are compared quantitatively on the problem of encoding the columns of multiple alignments. This generic problem has some attractive features:
The encoding cost (sometimes called conditional entropy) is a good measure of the variation among sequences of the multiple alignment. Since entropy is additive, the encoding cost for independent columns can be added to get the encoding cost for entire sequences, and strict significance tests can be applied by looking at the difference in encoding cost between a hypothesized model and a null model [16].
Each column t of a multiple alignment will give us a (possibly
weighted) count of amino acids, 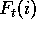 .
If we write the sum of all the counts for a column as
.
If we write the sum of all the counts for a column as 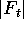 , we can
estimate the probability of each amino acid in the column as
, we can
estimate the probability of each amino acid in the column as
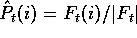 .
This is known as the maximum-likelihood estimate of the
probabilities.
Note: throughout this paper the notation |y| will mean
.
This is known as the maximum-likelihood estimate of the
probabilities.
Note: throughout this paper the notation |y| will mean
 for any vector of
values y.
for any vector of
values y.
To evaluate the regularizers, we want to see how accurately they predict the true probabilities of amino acids, given a sample. Unfortunately, true probabilities are never available with finite data sets. To avoid this problem, we will take a small sample of amino acids from the column, apply a regularizer to it, and see how well the regularizer estimates the maximum-likelihood probabilities for the whole column.
Let's use 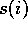 to be the number of occurrences of amino acid i in
the sample.
The estimated probability of amino acid i given the sample s will
be written as
to be the number of occurrences of amino acid i in
the sample.
The estimated probability of amino acid i given the sample s will
be written as 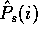 , and
the Shannon entropy or encoding cost of amino acid i given
the sample is
, and
the Shannon entropy or encoding cost of amino acid i given
the sample is 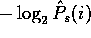 .
The average encoding cost for column t given sample s is the
weighted average over all
amino acids in the column of the encoding for that amino acid:
.
The average encoding cost for column t given sample s is the
weighted average over all
amino acids in the column of the encoding for that amino acid:
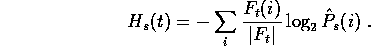
The better the estimation  is of
is of 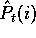 , the lower
the encoding cost
, the lower
the encoding cost 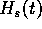 will be. The lowest possible value would
be obtained if the estimate were exact:
will be. The lowest possible value would
be obtained if the estimate were exact:
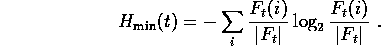
To make a fair comparison of regularizers, we should not look at a single sample s, but at the expected value when a sample of size k is chosen at random:
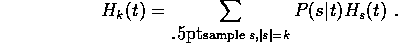
The weighting for each of the encoding costs 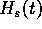 is the
probability of obtaining that particular sample from that column.
If the samples of size |s| are drawn by
independent selection with replacement from the density
is the
probability of obtaining that particular sample from that column.
If the samples of size |s| are drawn by
independent selection with replacement from the density 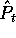 , then
the probability of each sample can be computed from the counts
, then
the probability of each sample can be computed from the counts 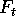 :
:
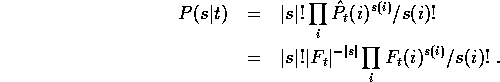
We can do a weighted average of the encoding costs over all columns to get the expected cost per amino acid for a given sample size:
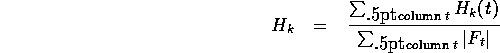
If we precompute the total count 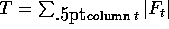 , and summary frequencies for each sample
, and summary frequencies for each sample
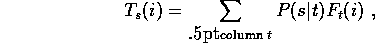
then we can rewrite the computation as
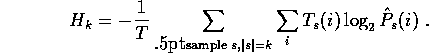
The average encoding cost 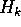 would be minimized if
would be minimized if
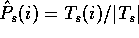 , giving us a lower bound on how well a
regularizer can do for samples of size k.
Table 1 shows this minimum average encoding cost
for the columns of the BLOCKS multiple
alignment [8] (with sequence weights explained in Section 4),
given that we have
sampled |s| amino acids from each column.
, giving us a lower bound on how well a
regularizer can do for samples of size k.
Table 1 shows this minimum average encoding cost
for the columns of the BLOCKS multiple
alignment [8] (with sequence weights explained in Section 4),
given that we have
sampled |s| amino acids from each column.
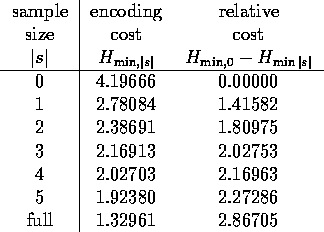
Table: The minimum
encoding cost of columns from the weighted BLOCKS
database, given that a sample of |s| amino acids is known.
The row labeled ``full'' is the cost if we know the
distribution for each column of the alignment
exactly, not just a sample from the column.
The last row of the table is the average encoding cost for the columns
if we use the full knowledge of the probabilities for the column
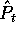 , rather than just a random sample.
This is the best we can hope to do with any method that treats the
columns independently.
It is probably not obtainable with any finite sample size, but we can
approach it if we use information other than just a sample of
amino acids to identify the column.
The relative entropy in the last column of
Table 1 measures how much information we have
gained by seeing a sample of |s| amino acids, relative to knowing
just the background probabilities.
, rather than just a random sample.
This is the best we can hope to do with any method that treats the
columns independently.
It is probably not obtainable with any finite sample size, but we can
approach it if we use information other than just a sample of
amino acids to identify the column.
The relative entropy in the last column of
Table 1 measures how much information we have
gained by seeing a sample of |s| amino acids, relative to knowing
just the background probabilities.
One disadvantage of the encoding cost computation used in this paper
is the cost of pre-computing the  values and computing the
values and computing the
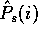 values for each of the possible samples.
The number of distinct samples to be examined is
values for each of the possible samples.
The number of distinct samples to be examined is 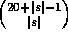 , which
grows exponentially with |s|, but remains manageable for
, which
grows exponentially with |s|, but remains manageable for 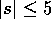 (42,504 distinct samples for |s|=5).
(42,504 distinct samples for |s|=5).