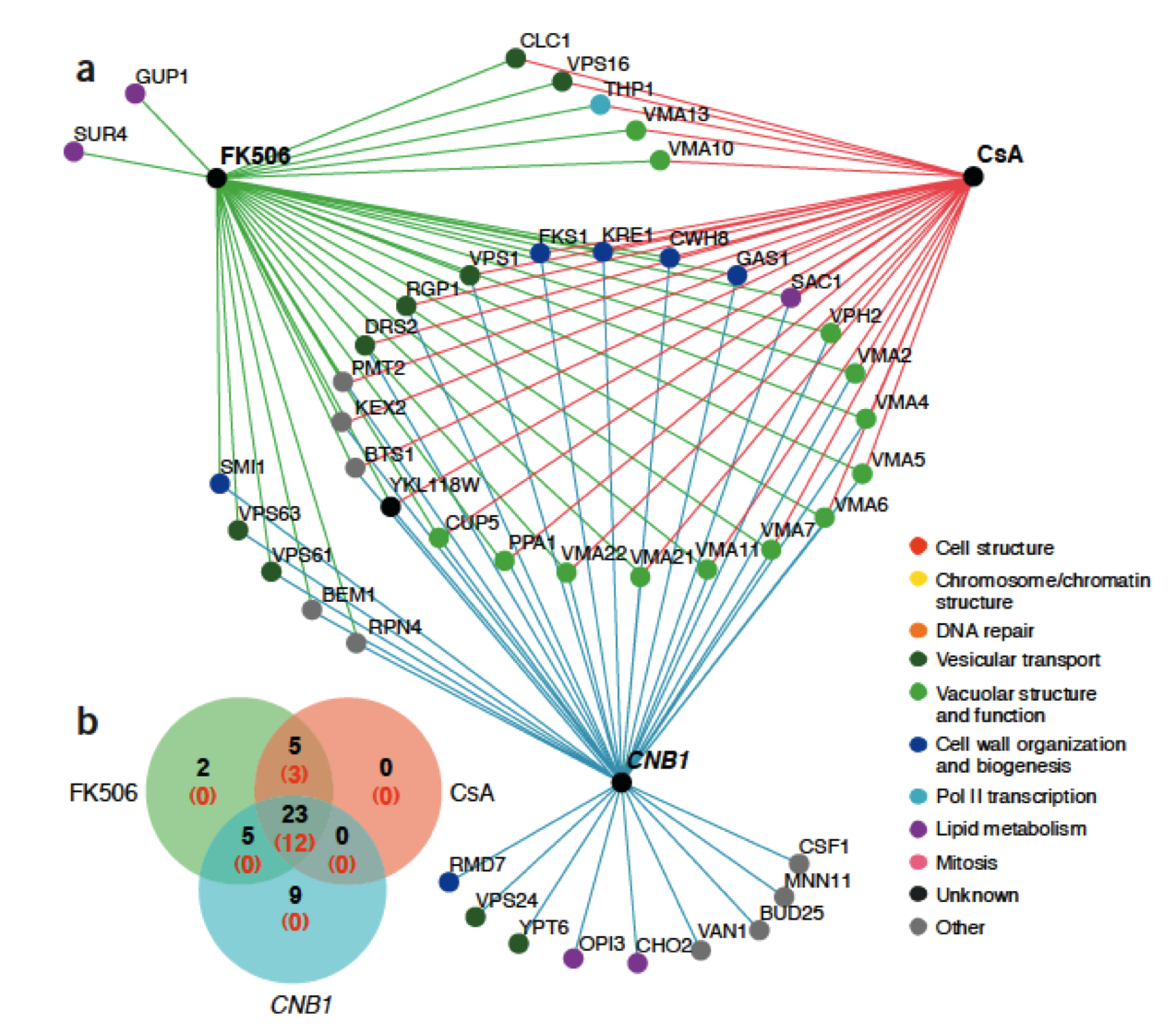
Genes act together to produce a phenotype for the cell. By studying the survivability of many combinations of double knockout mutants in yeast, the Boone lab and others have managed to construct an interaction map of a large percentage of non-vital genes in yeast (called a synthetic lethal map). Similar studies have constructed maps of chemical-genetic interactions by testing the survivability of yeast knockouts in chemical media. Using informatics, these data can be combined to predict the genetic and pathway targets of chemical compounds in yeast as a model organism for humans.
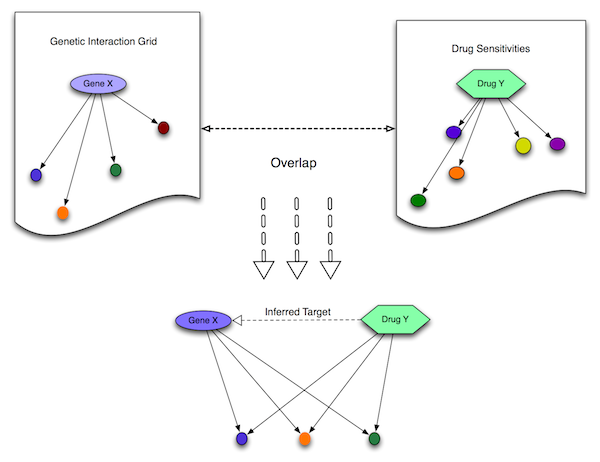
My first task was to build a software pipeline to take any given set of experimental chemical-genomics data and produce a list of drug target predictions using a genetic synthetic lethal map. The algorithm first takes each vector of chemical-genetic interaction values and thresholds it to produce a pared list of genes that interact with that chemical -- the chemical’s gene set. These gene sets are then overlapped with the genetic synthetic lethal sets in one of a variety of tunable ways -- hypergeometric overlap, a KS test, or Euclidean distance. The genes which overlap most significantly with each chemical’s gene set are considered the target picks.
Multi drug resistant genes are those genes that protect the cell from a variety of chemical compounds, and will therefore show interactions with a number of chemical compounds in a given chemical-genomic experiment. These genes will appear as false positives for many of the chemical compounds, and need to be removed from the experimental data before running the algorithm. The simplest approach is simply to produce an ordered list of genes and eliminate those which interact with more than an arbitrary number of compounds; but there are better techniques to find these genes.
I implemented an algorithm that finds MDR genes by using the mutual information provided by the entire set of genes in a given chemical-genomic dataset. The efficacy of this pre-processing step was then tested by comparing the target predictions on known compounds with the predictions using the naive approach.
I evaluated the performance of the algorithm and the parameters by having it predict the targets of known chemical compounds, and comparing the predictions with the targets curated by biologists at USC. The evaluation was performed by running a gene set enrichment test for enrichment of the curated genes against the algorithm’s prediction distribution, for each chemical. The GSEA score for each chemical was summed to produce a final ‘score’ for the algorithm’s efficacy. Compounds were sorted according to the number of high-confidence predictions produced for each (-log10 pvalue greater than 5).