One of the projects I worked on involved testing a software package callled the Pairwise Sequentially Markovian Coalescent Model(PSMC). I was tasked with determining if you could infer ancient bottleneck events using this model.
One of the reasons we are interested in studying changes in the ancestral population of a species is that it allows us to infer important events in the history of that species such as speciation, migration and the formation of subpopulations. In addition, it allows us to make inferences about the fitness of that species and how that fitness has changed alongside changes in the climate and the emergence of competeing species. One of the tools used to study ancestral population size is a program called PSMC. PSMC is a hidden markov model that uses a diploid genome as its input and returns a table or graph telling you the population size of that organisms ancestors at different points in time.
PSMC does this by measuring heterozygosity between the two copies of a chromosome. If you know the mutation and recombination rate for a species, you can infer the time in the past when a given segment of DNA shared a common ancestor. By measuring the coalescence time across the entire genome you can infer the population size at various points in time because coalescence time increases with population size.
Basically, the more individuals you have in a population, the more heterozygosity you get in your genome. whereas fewer individuals will produce less heterozygosity.
In order to test the program's ability to infer ancestral bottleneck events, a script was created to generate simulated genomes using different parameters such as the duration, intensity and time at which a bottleneck occured in that organism's ancestery. The infered population was compared to the actual population level.
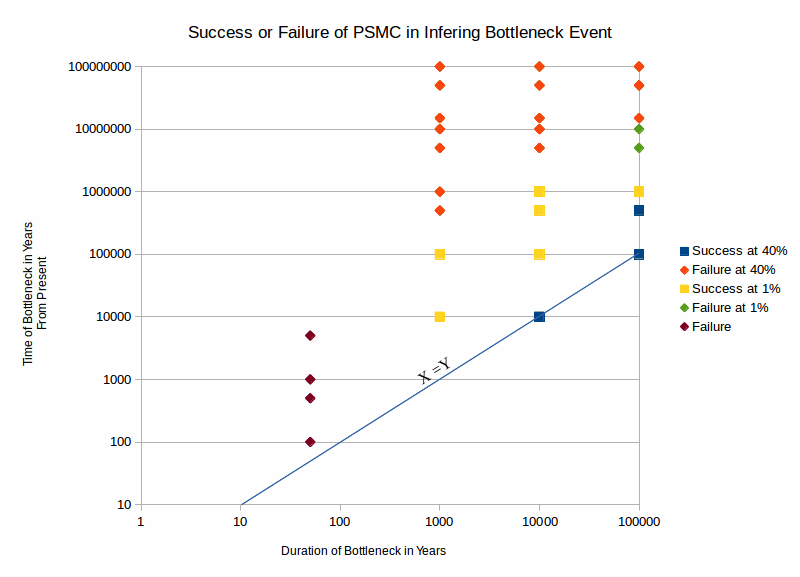
This graph shows where PSMC succeeded or failed when the magnitude of the bottleneck event was fixed. The conclusions I derived from these simulations are as follows:
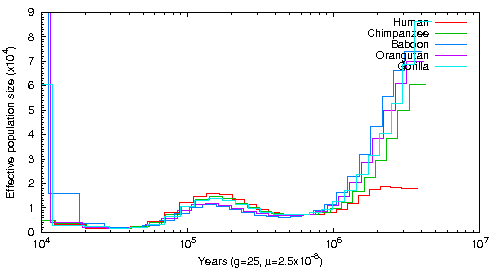
Another project I worked on in Beth Shapiro's lab involved testing the capabilities of PSMC to see if it was possible to infer ancestral population size by aligning sequence data to the genomes of distantly related species. The lab studies the genomes of extinct and exotic species for which no high-quality reference genomes exist. Theoretically, one could infer ancestral population sizes from these species if you aligned short-read data to a sufficiently close relative. However, the more divergent a genome is the more difficult it becomes to align sequences and to, consequently, infer heterozygous sites. In order to test PSMCs capabilities, I aligned Human read data to Human, Chimpanzee, Gorilla, Baboon and Orangutan genomes and trained PSMC using these different alignments. This figure shows the result of estimating the population based on these alignments using one set of SNP estimation parameters. This figure shows the result using a different set of SNP estimation parameters. The conclusion is that it is possible to infer population size using closely related species as long as SNP calling is done carefully.
This graph was created aligning Human short read data to Human, Chimpanzee, Orangutan, Gorilla and Baboon Genomes. The alignment tool was BWA and the SNPs were called with Samtools mpileup without the -C 50 command. The exact script can be found here.
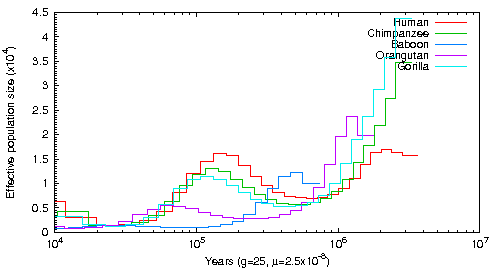
This graph was created aligning Human short read data to Human, Chimpanzee, Orangutan, Gorilla and Baboon Genomes. The alignment tool was BWA and the SNPs were called with Samtools mpileup with the -C 50 command. The exact script can be found here.
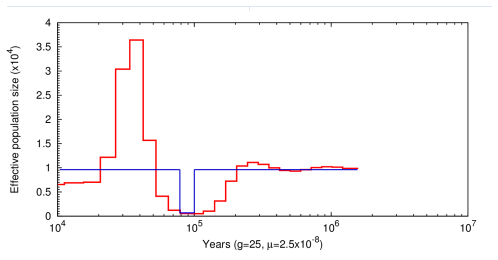
This is an example of the output produced by PSMC. The blue line is the actual population history and the red line represnts the infered population. This would be considered a successful inference by PSMC.