I wrote my own code for this before the solution was given:
cocluster.index <- function (a, b) {
match <- mismatch <- 0
for (i in 1:length(a)) {
apairs <- which(a == a[i])
bpairs <- which(b == b[i])
apairs <- apairs[apairs>i]
bpairs <- bpairs[bpairs>i]
o <- which(apairs %in% bpairs)
overlap <- length(o)
match <- match+overlap
mismatch <- mismatch + length(apairs) + length(bpairs) - 2*overlap
}
match / (match+mismatch)
}centers <- 15
runs <- 10
cluster.kmeans <- apply(as.matrix(1:runs), 1,
function(x){kmeans(stress.submatrix,centers)$cluster})
cluster.kmeans.cocluster <- NULL
for(i in 1:(runs-1)) {
for (j in (i+1):runs) {
val <- cocluster.index(cluster.kmeans[,i],cluster.kmeans[,j])
cluster.kmeans.cocluster <- c(cluster.kmeans.cocluster, val)
}
}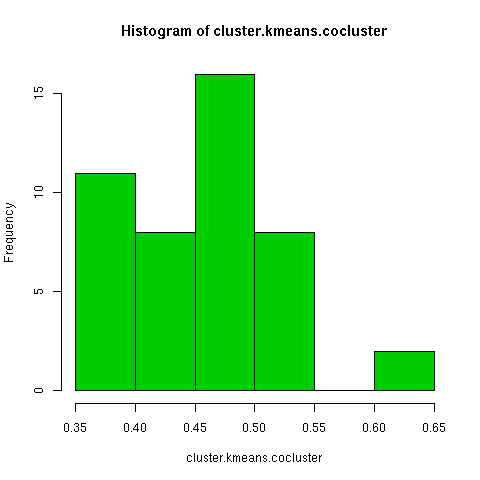
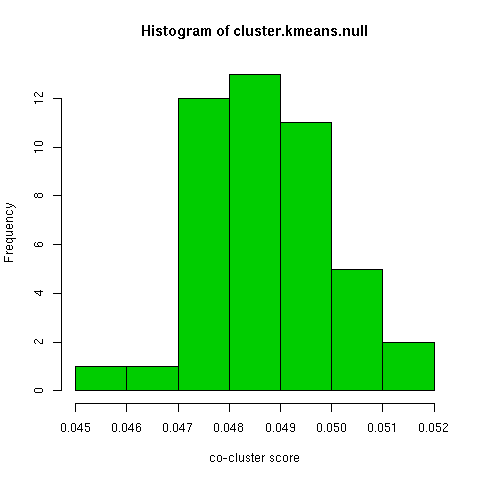
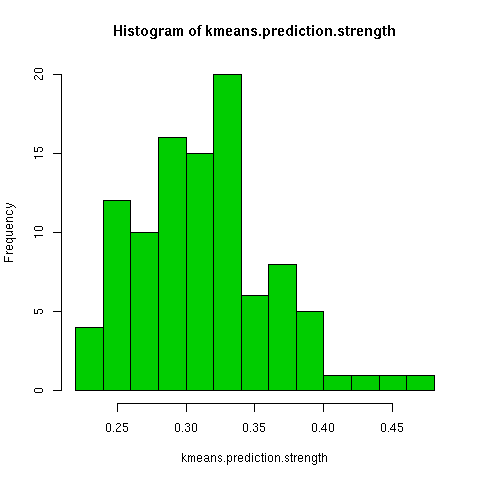
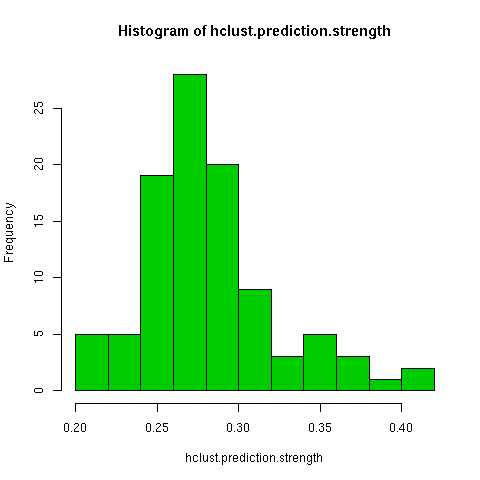
The kmeans clustering has a wide degree of variability under this measure, about 0.3. When i randomly permuted the first clustering and used the co-cluster measure, the range of scores was much smaller than between all the kmeans clusterings, by about a third. For a more direct comparison with the permuted co-clusters, I looked at just the comparison of the first kmeans clustering with the other 9 kmeans clusterings, and this distribution had very little overlap with the null distribution.
By trying kmeans-clustering 10 times on each value of k, for lots of values of k, you could pick the k that gives you the smallest mean cocluster.index value.