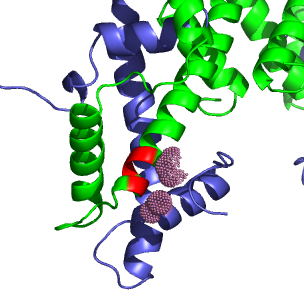
H3.3 variations. H3 is shown in green, H4 in blue. The locations of variations in H3.3 are shown red, and the atoms of H4 within 5Å of the variable H3/H3.3 residues are shown in pink.
For this problem, I examined H3 and H3.3 in fly. However, the crystal structure was solved with xenopus histones, so I used a multiple alignment to find all differences.
Fly_H3 MARTKQTARKSTGGKAPRKQLATKAARKSAPATGGVKKPHRYRPGTVALREIRRYQKSTE
Fly_H3.3 MARTKQTARKSTGGKAPRKQLATKAARKSAPSTGGVKKPHRYRPGTVALREIRRYQKSTE
Xenopus_H3 MARTKQTARKSTGGKAPRKQLATKAARKSAPATGGVKKPHRYRPGTVALREIRRYQKSTE
PDB --------------------LATKAARKSAPATGGVKKPHRYRPGTVALREIRRYQKSTE
***********:****************************
Fly_H3 LLIRKLPFQRLVREIAQDFKTDLRFQSSAVMALQEASEAYLVGLFEDTNLCAIHAKRVTI
Fly_H3.3 LLIRKLPFQRLVREIAQDFKTDLRFQSAAIGALQEASEAYLVGLFEDTNLCAIHAKRVTI
Xenopus_H3 LLIRKLPFQRLVREIAQDFKTDLRFQSSAVMALQEASEAYLVGLFEDTNLCAIHAKRVTI
PDB LLIRKLPFQRLVREIAQDFKTDLRFQSSAVMALQEASEAYLVALFEDTNLCAIHAKRVTI
***************************:*: ***********.*****************
Fly_H3 MPKDIQLARRIRGERA
Fly_H3.3 MPKDIQLARRIRGERA
Xenopus_H3 MPKDIQLARRIRGERA
PDB MPKDIQLARRIRGERA
****************
|
H3.3 variations. H3 is shown in green, H4 in blue. The locations of variations in H3.3 are shown red, and the atoms of H4 within 5Å of the variable H3/H3.3 residues are shown in pink. |
Three are four differences between H3 and H3.3:
The difference at position 32 is in the N-terminal coil, and in Luger's crystal structure is located in solution just outside the DNA loops. Any variation in H3.3's function due to this residue would have to involve a factor that binds very close to nucleosome. The other three different residues are towards the N-terminus of the large alpha-helix. The only neighboring atoms within 5Å are on H4 and H3 itself. On H4, there are only Ala84 and Phe101, both of which are nonpolar, and will interact favorably with all the H3.3 variations. Ser88 in H3 does not appear to interact with H4. There seem to be no changes in polar interactions between between the H3-H4 tetramer and H3.3-H4 tetramer.
Lys65, Phe68, Gln69, Val72, and Arg73 of H3 are within 5Å of the helix that abuts the variant residue positions, and of the variant residue positions, only Val91 has a side chain that interacts with the smaller helix. A substitution of isoleucine for valine will have little affect on the interaction of the two helices.
All of the internal changes between H3.3 and H3 seem unlikely to significantly change the structure except for the inclusion of a glycine. Glycine is rarely found in a helix conformations, so in H3.3, this substitution will either break or at least destabilize it's containing helix. This could lead to greater conformational freedom or slightly different shape and DNA binding affinities when comparing H3.3 to H3.
The residue differences do not provide much of a clue to H3.3's purpose, but its localization might. If H3.3 nucleosomes were easier (either in energy or required time) to remodel, then they would be better suited to positioning over heavily transcribed genes, as their localization indicates.
Though in vitro experiments are not definitive for modeling some aspects of chromatin, I believe that they are helpful for testing the biochemical properties of proteins and compounds. I hypothesize that H3.3 nucleosomes are "easier" to remodel than H3 nucleosomes due differing biochemical properties, and an in vitro assay should be sufficient to test this hypothesis.
My biochemical assay would be based on the assays used to investigate RSC (Saha, 2002. Genes and Dev.). I would assemble separate samples of mononucleosomes with H3 and the H3.3 variant. The mononucleosomes would be wrapped with DNA that contains the DraI restriction site. Both species of mononucleosomes would be incubated with the RSC complex, ATP, and the DraI restriction enzyme. Remodeled nucleosomes would free the DNA to be cut by DraI, and by labeling the DNA and running it on a gel, the relative proportion of cut to uncut DNA can be measured, and therefore the proportion of remodeled to undisturbed nucleosomes. Measuring the amount of hydrolyzed ATP will give the amount of energy consumed, and dividing the amount of cut DNA by the amount of hydrolyzed ATP will give an indication of the energy required to remodel an H3 or H3.3 mononucleosome.
I favor a model where histones are fully displaced as transcription occurs, and then replaced after RNA polymerase has passed. During S phase, H3.3 expression is suppressed, and H3 expression is enhanced, resulting in packaging of primarily H3 nucleosomes. In other cell phases, H3.3 would be up-regulated, and H3 down-regulated. As H3 nucleosomes are removed from DNA by the transcription process they are replaced by the more abundant H3.3 nucleosomes, accounting for the presence of H3.3 at transcriptionally active loci.
The most common model for cis-displacement of nucleosomes by chromatin remodellers involves the creation of bulge of DNA along a histone through twisting of the DNA. The bulge would then propagate by diffusion. However, there are some objections to this model, as nicked DNA does not greatly affect the chromatin remodeling (Aoyagi, et al, 2003). In addition, Pol II transcription results in considerable disruption of the chromatin structure, removing an H2A/H2B dimer (Kireeva, 2002). Therefore, I think there is room for another model of chromatin remodeling during transcription, in which histones can be completely removed from DNA. This is supported by data in from experiments involving the deposition of H3.3 nucleosomes (Ahmad and Henikoff, 2002).
One of the first controls on the experiment should test to see that the antibody is specific to phosphorylated H1 and does not bind other proteins in the cell. Of particular concern for this experiment are DNA binding proteins and proteins that bind to DNA binding proteins. To control for this, I would dissociate protein complexes in the cell extract for the ChIP experiments, remove the DNA, and then run that through my antibody column. After eluting the bound protein, I would run that on a 2D gel to check that only one species bound to the antibody, and that it had the weight and pH of H1. This protein should also be assayed for the presence of phosphorylation on Ser53.
In addition, other transcriptionally active and inactive sites should be queried before the association of Ser53-phosphorylated H1 can be generalized. H1 phosphorylation might be specific to PMA1, or the phosphorylation might be distributed without regard to transcription.
It would also be preferable to associate H1 Ser53-phosphorylation with other transcriptionally-active modifications to histones. This could be achieved by running the first ChIP on only lightly digested chromatin (to insure that H1 is still linked to nearby nucleosomes), and then running another ChIP serially that will probe for H3 or H4 acetylation.